Assessment |
Biopsychology |
Comparative |
Cognitive |
Developmental |
Language |
Individual differences |
Personality |
Philosophy |
Social |
Methods |
Statistics |
Clinical |
Educational |
Industrial |
Professional items |
World psychology |
Clinical: Approaches · Group therapy · Techniques · Types of problem · Areas of specialism · Taxonomies · Therapeutic issues · Modes of delivery · Model translation project · Personal experiences ·
- This article is about a type of spinal muscular atrophy linked to a genetic defect in the AR gene. For a list of other conditions with similar names, see Spinal muscular atrophies.
| ICD-10 | G121 | |
|---|---|---|
| ICD-9 | 335.1 | |
| OMIM | 313200 | |
| DiseasesDB | 7144 | |
| MedlinePlus | [1] | |
| eMedicine | article/1172604 | |
| MeSH | {{{MeshNumber}}} | |
Spinal and bulbar muscular atrophy (SBMA), also known as spinobulbar muscular atrophy, bulbo-spinal atrophy, X-linked bulbospinal neuropathy (XBSN), X-linked spinal muscular atrophy type 1 (SMAX1), and Kennedy's disease (KD) — is a form of atrophy and is a debilitating neurodegenerative disease resulting in muscle cramps and progressive weakness due to degeneration of motor neurons in the brain stem and spinal cord.
The condition is associated with mutation of the androgen receptor (AR) gene[1][2][3] and is inherited in a X-linked recessive manner. As with many genetic disorders, no cure is known, although research continues.
Because of its endocrine manifestations related to the impairment of the AR gene, SMBA can be viewed as a variation of the disorders of the androgen insensitivity syndrome (AIS). It is also related to other neurodegenerative diseases caused by similar mutations, such as Huntington's disease and the spinocerebellar ataxias.
Genetics[]
{kind=link}
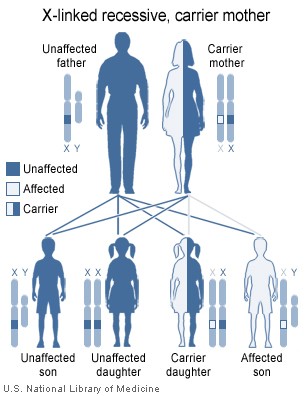
SBMA is inherited in an X-linked recessive pattern.
The androgen receptor gene that is mutated in SBMA is located on the X chromosome, and the effects of the mutation may be androgen-dependent, thus only males are fully affected. Females are rarely affected; female carriers tend to have a relatively mild expression of the disease if they show symptoms at all.
Please note that the diagrams will be different if 1) the father is affected AND the mother is unaffected OR 2) if the father is affected and the mother is a carrier.
Pathophysiology[]
As reported in 1991, SBMA is caused by expansion of a CAG repeat in the first exon of the androgen receptor gene (trinucleotide repeats).[4] The CAG repeat encodes a polyglutamine tract in the androgen receptor protein. The greater the expansion of the CAG repeat, the earlier the disease onset and more severe the disease manifestations. The repeat expansion likely causes a toxic gain of function in the receptor protein, since loss of receptor function in androgen insensitivity syndrome does not cause motor neuron degeneration. Spinal and bulbar muscular atrophy may share mechanistic features with other disorders that are caused by polyglutamine expansion, such as Huntington's disease. There is currently no treatment or cure for SBMA.
It is a lower motor neuron disease.[5]
Signs and symptoms[]
SBMA patients have muscle cramps and progressive weakness due to degeneration of motor neurons in the brain stem and spinal cord.
Ages of onset and severity of manifestations in affected males vary from adolescence to old age, but most commonly develop in middle adult life. The latest onset was described in a male of 84 years of age. KD does not usually compromise longevity. The syndrome has neuromuscular and endocrine manifestations.
Neuromuscular[]
Early signs often include weakness of tongue and mouth muscles, fasciculations, and gradually increasing weakness of limb muscles with muscle wasting. In some cases, premature muscle fatigue begins in adolescence. Neuromuscular management is supportive, and the disease progresses very slowly but can eventually lead to extreme disability.
Neurological:
- Bulbar signs: The bulbar muscles are those supplied by the motor nerves from the brain stem, which control swallowing, breathing, speech, and other functions of the throat. Bulbar signs are problems with these muscles.
- Lower motor neuron signs: The lower motor neurons are those in the brainstem and spinal cord that directly supply the muscles. Loss of lower motor neurons leads to weakness and wasting of the muscle.
- Primary sensory neuropathy: Loss of sensation and numbness, usually not noticeable.
- Intention tremor: Hand tremor with volitional effort.
- Babinski (plantar) response: When the bottom of the foot is scraped, the toes bend down. An abnormal response would be an upward movement of the toes indicating a problem with higher level (upper) motor neurons.
- Decreased or absent deep tendon reflexes: When a doctor taps the knee with his hammer little or nothing happens.
Muscular:
- Fasciculations: Twitching of muscles when at rest.
- Cramps: Large muscle spasms.
- Muscular atrophy: Loss of muscle bulk that occurs when the lower motor neurons do not stimulate the muscle adequately.
Endocrine
- Gynecomastia: breast enlargement.
- Impotence
- Erectile dysfunction
- Reduced fertility
- Low sperm count
- Testicular atrophy: Testicles become smaller and less functional.
Miscellaneous Characteristics:
- Late onset: Patients usually develop symptoms in the late 30's or later
- Symmetry of clinical signs: Muscles are usually affected symmetrically.
Homozygous females[]
Homozygous females, both of whose X chromosomes have a mutation leading to CAG expansion of the AR gene, have been reported to show only mild symptoms of muscle cramps and twitching. No endocrinopathy has been described.
History[]
This disorder was first described by Dr. William R. Kennedy in 1968.[6] In 1991 it was recognized that the AR gene is involved in the disease process. The disease is probably more common than originally thought. A study in Scandinavia suggested a prevalence of 1.3/8,500 making KD the most common form of motor neuron disease in the specific area studied; nobody had been diagnosed before 1995. It has been suggested that some men with SBMA may be misdiagnosed to have amyotrophic lateral sclerosis (ALS).
See also[]
References[]
- ↑ DOI:10.1016/S1047-9651(02)00119-5
This citation will be automatically completed in the next few minutes. You can jump the queue or expand by hand - ↑ DOI:10.1098/rstb.1999.0461
This citation will be automatically completed in the next few minutes. You can jump the queue or expand by hand - ↑ Chen CJ, Fischbeck KH (2006). "Ch. 13: Clinical aspects and the genetic and molecular biology of Kennedy's disease" Tetsuo Ashizawa; Wells, Robert V. Genetic Instabilities and Neurological Diseases, 2nd, 211–222, Boston: Academic Press.
- ↑ DOI:10.1038/352077a0
This citation will be automatically completed in the next few minutes. You can jump the queue or expand by hand - ↑ DOI:10.1602/neurorx.2.3.471
This citation will be automatically completed in the next few minutes. You can jump the queue or expand by hand - ↑ PMID 4233749 (PMID &query_hl=14&itool=pubmed_docsum 4233749 )
Citation will be completed automatically in a few minutes. Jump the queue or expand by hand
|}
Sex linkage: X-linked recessive disorders | |
|---|---|
| Immune |
Chronic granulomatous disease (CYBB) · Wiskott-Aldrich syndrome · X-linked Severe Combined Immunodeficiency · X-linked agammaglobulinemia · Hyper-IgM syndrome type 1 · IPEX |
| Hematologic |
Haemophilia A · Haemophilia B · X-linked sideroblastic anemia · X-linked lymphoproliferative disease |
| Endocrine |
Androgen insensitivity syndrome/Kennedy disease · Diabetes insipidus |
| Metabolic |
amino acid: Ornithine transcarbamylase deficiency · Oculocerebrorenal syndrome dyslipidemia: Adrenoleukodystrophy carbohydrate metabolism: Glucose-6-phosphate dehydrogenase deficiency · Pyruvate dehydrogenase deficiency · Danon disease/glycogen storage disease Type IIb lipid storage disorder: Fabry's disease mucopolysaccharidosis: Hunter syndrome purine-pyrimidine metabolism: Lesch-Nyhan syndrome |
| Nervous system |
X-Linked mental retardation: Coffin-Lowry syndrome · Fragile X syndrome · MASA syndrome · Rett syndrome eye disorders: Color blindness (red and green, but not blue) · Ocular albinism (1) · Norrie disease · Choroideremia other: Charcot-Marie-Tooth disease (CMTX2-3) · Pelizaeus-Merzbacher disease |
| Skin |
Dyskeratosis congenita · Hypohidrotic ectodermal dysplasia (EDA) · X-linked ichthyosis |
| Neuromuscular |
Becker's muscular dystrophy/Duchenne · Centronuclear myopathy · Myotubular myopathy · Conradi-Hünermann syndrome |
| Urologic |
Alport syndrome · Dent's disease |
| No primary system |
Barth syndrome · McLeod syndrome · Simpson-Golabi-Behmel syndrome |
|
Note: there are very few X-linked dominant disorders. These include X-linked hypophosphatemia, Focal dermal hypoplasia, Aicardi syndrome, Incontinentia pigmenti, and CHILD. | |
Template:Trinucleotide repeat disorders Template:Intracellular receptor deficiencies
| This page uses Creative Commons Licensed content from Wikipedia (view authors). |