Assessment |
Biopsychology |
Comparative |
Cognitive |
Developmental |
Language |
Individual differences |
Personality |
Philosophy |
Social |
Methods |
Statistics |
Clinical |
Educational |
Industrial |
Professional items |
World psychology |
Clinical: Approaches · Group therapy · Techniques · Types of problem · Areas of specialism · Taxonomies · Therapeutic issues · Modes of delivery · Model translation project · Personal experiences ·
Philip_Baker.jpg|
| ICD-10 | E791 | |
|---|---|---|
| ICD-9 | 277.2 | |
| OMIM | 308000 | |
| DiseasesDB | 7415 | |
| MedlinePlus | 001655 | |
| eMedicine | neuro/630 | |
| MeSH | {{{MeshNumber}}} | |
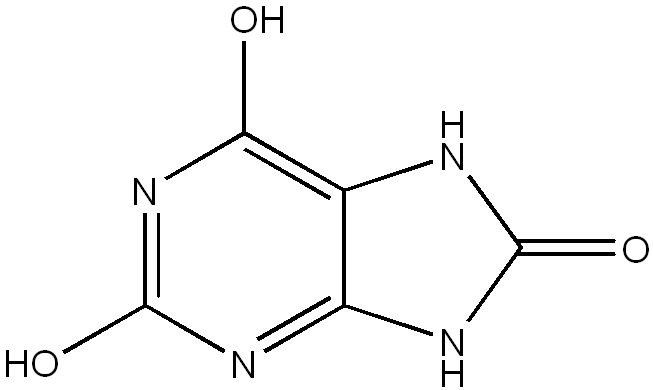
Lesch-Nyhan syndrome (LNS) is a rare, inherited disorder caused by a deficiency of the enzyme hypoxanthine-guanine phosphoribosyltransferase (HPRT). LNS is an X-linked recessive disease: the gene is carried by the mother and passed on to her son. LNS is present at birth in babies.
Patients have severe mental and physical problems throughout life. The lack of HPRT causes a build-up of uric acid in all body fluids, and leads to problems such as severe gout, poor muscle control, and moderate intellectual disability, which appear in the first year of life. A striking feature of LNS is self-injurious behaviors, characterized by lip and finger biting, that begin in the second year of life.
Abnormally high uric acid levels can cause sodium urate crystals to form in the joints, kidneys, central nervous system and other tissues of the body, leading to gout-like swelling in the joints and severe kidney problems. Neurological symptoms include facial grimacing, involuntary writhing, and repetitive movements of the arms and legs similar to those seen in Huntington's disease. The direct cause of the neurological abnormalities remains unknown. Because a lack of HPRT causes the body to poorly utilize vitamin B12, some boys may develop a rare disorder called megaloblastic anemia.
The symptoms caused by the buildup of uric acid (arthritis and renal symptoms) respond well to treatment with drugs such as allopurinol that reduce the levels of uric acid in the blood. The mental deficits and self-mutilating behavior do not respond to treatment. There is no cure, but many patients live to adulthood. LNS is rare, affecting about one in 380,000 live births. It was first described in 1964 by Dr. Michael Lesch and Dr. William Nyhan.
Features[]
LNS is characterized by three major hallmarks: neurologic dysfunction, cognitive and behavioral disturbances, as well as uric acid overproduction (hyperuricemia). Some may also be afflicted with anemia (macrocytic). Virtually all patients are male, and male victims suffer delayed growth and puberty, and most develop shrunken testicles or testicular atrophy. Female carriers are at an increased risk for gouty arthritis, but are usually otherwise unaffected.
Overproduction of uric acid[]
One of the first symptoms of the disease is the presence of sand-like crystals of uric acid in the diapers of the affected infant. Overproduction of uric acid may lead to the development of uric acid crystals or stones in the kidneys, ureters, or bladder. Such crystals deposited in joints later in the disease may produce gout-like arthritis, with swelling and tenderness.
{kind=link}
uric acid molecule
The overproduction of uric acid is present at birth, but may not be recognized by routine clinical laboratory testing methods. The serum uric acid concentration is often normal, as the excess purines are promptly eliminated in the urine. The crystals usually appear as an orange grainy material, or they may coalesce to form either multiple tiny stones, or distinct large stones that are difficult to pass. The stones, or calculi, usually cause hematuria (blood in the urine) and increase the risk of urinary tract infection. Some victims suffer kidney damage due to such kidney stones. Stones may be the presenting feature of the disease, but can go undetected for months or even years.
Nervous system impairment[]
The periods before and surrounding birth are typically normal in individuals with LNS. The most common presenting features are abnormally decreased muscle tone (hypotonia) and developmental delay, which are evident by three to six months of age. Affected individuals are late in sitting up, while most never crawl or walk. Lack of speech is also a very common trait associated with LNS.
Irritability is most often noticed along with the first signs of nervous system impairment. Within the first few years of life, extrapyramidal involvement causes abnormal involuntary muscle contractions such as loss of motor control (dystonia), writhing motions (choreoathetosis), and arching of the spine (opisthotonus). Signs of pyramidal system involvement, including spasticity, overactive reflexes (hyperreflexia) and extensor plantar reflexes, also occur. The resemblance to athetoid cerebral palsy is apparent in the neurologic aspects of LNS. As a result, most individuals are initially diagnosed as having cerebral palsy. The motor disability is so extensive that most individuals never walk, and are confined to a wheelchair for life.
Self-injuring behavior[]
Persons affected are cognitively impaired and have behavioral disturbances that emerge between two and three years of age. The uncontrollable self-injury associated with LNS also usually begins at three years of age. The self-injury begins with biting of the lips and tongue and as the disease progresses, affected individuals frequently develop finger biting and head banging. The self-injury can increase during times of stress. Self-mutilation is a distinguishing characteristic of the disease and is apparent in 85% of affected males.
The majority of individuals are cognitively impaired, which is not easy to determine because of the behavioral disturbances and motor deficits associated with the syndrome. In many ways, the behaviors may be seen as a psychological extension of the compulsion to cause self-injury: rejecting desired treats or travel, repaying kindness with coldness or rage, failing to answer test questions correctly despite study and a desire to succeed, provoking anger from caregivers when affection is desired, and so on.
Compulsive behaviors also occur, including aggressiveness, vomiting, spitting, and involuntary swearing, or coprolalia. The development of this type of behavior is sometimes seen within the first year, or in early childhood, but others may not develop it until later in life.
LNS in females[]
While carrier females are generally asymptomatic, they do experience an increase in uric acid excretion, and some may develop symptoms of hyperuricemia, and suffer from gout in their later years. Testing in this context has no clinical consequence, but it may reveal the possibility of transmitting the trait to male children. Women may also require testing if a male child develops LNS. In this instance, a negative test means the son's disease is the result of a new mutation, and the risk in siblings is not increased.
Females who carry one copy of the defective gene are asymptomatic carriers with a 50% chance of passing the disease on to their sons. In order for a female to be affected, she would need to have two copies of the mutated gene, one of which would be inherited from her father. Males affected with LNS do not usually have children due to the debilitating effects of the disease. It is possible for a female to inherit an X chromosome from her unaffected father, who carries a new mutation of the HPRT gene. Under these circumstances, a girl could be born with LNS, and there are a few reports of this happening, but it is very rare. The overwhelming majority of patients with LNS are male.
Diagnosis[]
When an affected individual has fully developed the three clinical elements of uric acid overproduction, neurologic dysfunction, and cognitive and behavioral disturbances, diagnosis of LNS is easily made. Difficulties of diagnosis are abundant in the early stages when the three features are not yet obvious. Suspicion often comes about when the developmental delay of the individual is associated with hyperuricemia. Otherwise, the diagnosis should be alleged when developmental delay is associated with kidney stones (nephrolithiasis) or blood in the urine (hematuria), caused by uric acid stones.
For the most part, Lesch-Nyhan syndrome is first suspected when self-injurious behavior develops. However, self-injurious behaviors occur in other conditions, including nonspecific intellectual disability, autism, Rett syndrome, Cornelia de Lange syndrome, Tourette syndrome, familial dysautonomia, choreoacanthocytosis, sensory neuropathy including hereditary sensory neuropathy type 1, and several psychiatric conditions.
Of these, only individuals with Lesch-Nyhan syndrome, de Lange syndrome, and familial dysautonomia recurrently display loss of tissue as a consequence. Biting the fingers and lips is a definitive feature of Lesch-Nyhan syndrome; in other syndromes associated with self-injury, the behaviors usually consist of head banging and nonspecific self-mutilation, but not biting of the cheeks, lips and fingers. Lesch-Nyhan syndrome ought to be clearly considered only when self-injurious behavior takes place in conjunction with hyperuricemia and neurological dysfunction.
Diagnostic approach[]
The urate to creatinine (breakdown product of creatine phosphate in muscle) concentration ratio in urine is elevated. This is a good indicator of acid overproduction. For children under ten years of age with Lesch-Nyhan syndrome, a urate to creatinine ratio above two is typically found. Twenty-four-hour urate excretion of more than 20 mg/kg is also typical but is not diagnostic. Hyperuricemia (serum uric acid concentration of >8 mg/dL) is often present but not reliable enough for diagnosis. Activity of the HPRT enzyme in cells from any type of tissue (e.g., blood, cultured fibroblasts, or lymphoblasts) that is less than 1.5% of normal enzyme activity confirms the diagnosis of Lesch-Nyhan syndrome.
Testing[]
The use of biochemical testing for the detection of carriers is technically demanding and not often used. Biochemical analyses that have been performed on hair bulbs from at risk women have had a small number of both false positive and false negative outcomes. If only a suspected carrier female is available for HPRT1 mutation testing, it is appropriate to grow her lymphocytes in 6-thioguanine (a purine analogue), which allows only HPRT-deficient cells to survive. A mutant frequency of 0.5-5.0 x 10-2 is found in carrier females, while a non-carrier female has a frequency of 1-20 x 10-6. This frequency is usually diagnostic by itself.
Molecular genetic testing is the most effective method of testing, as HPRT1 is the only gene known to be associated with LNS. Individuals who display the full Lesch-Nyhan phenotype all have mutations in the HPRT1 gene. Sequence analysis of mRNA is available clinically and can be utilized in order to detect HPRT1 mutations in males affected with Lesch-Nyhan syndrome. Techniques such as RT-PCR, multiplex genomic PCR, and sequence analysis (cDNA and genomic DNA), used for the diagnosis of genetic diseases, are performed on a research basis. If RT-PCR tests result in cDNA showing the absence of an entire exon or exons, then multiplex genomic PCR testing is performed. Multiplex genomic PCR testing amplifies the nine exons of the HPRT1 gene as eight PCR products. If the exon in question is deleted, the corresponding band will be missing from the multiplex PCR. However if the exon is present, the exon is sequenced to identify the mutation, therefore causing exclusion of the exon from cDNA. If no cDNA is created by RT-PCR, then multiplex PCR is performed on the notion that most or all, of the gene is obliterated.
Genetics[]
{kind=link}
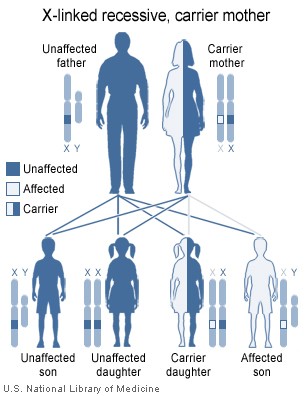
LNS is inherited in an X-linked recessive fashion.
LNS is due to mutations in the HPRT1 gene, so named because it codes for the enzyme hypoxanthine-guanine phosphoribosyltransferase (HPRT or HGPRT, EC 2.4.2.8). This enzyme is involved in the biochemical pathways the body uses to produce purines, one of the components of DNA and RNA. Defects of this enzyme lead to increased production of uric acid. Since the HPRT gene is located on the X chromosome, LNS is an X-linked inherited disease.
The father of an affected male will not be the carrier of the mutant allele, and will not have the disease. An obligate carrier would be a woman who has an affected son and one other affected relative in the maternal line.
If a woman is the first in her family with an affected son, Haldane's rule predicts a 2/3 chance that she is a carrier and a 1/3 chance that the son has a new germline mutation. However, in this case Haldane's prediction is incorrect due to an increased risk of mutation arising from the father when compared to the mother.
The risk to siblings of an affected individual depends upon the carrier status of the mother herself. A 50% chance is given to any female who is a carrier to transmit the HPRT1 mutation in each pregnancy. Sons who inherit the mutation will be affected while daughters who inherit the mutation are carriers. Therefore, with each pregnancy, a carrier female has a 25% chance of having a male that is affected, a 25% chance of having a female that is a carrier, and a 50% chance of having a normal male or female.
Males with LNS do not reproduce due to the characteristics of the disease. However, if a male with a less severe phenotype reproduces, all of his daughters are carriers, and none of his sons will be affected.
Pathophysiology[]
As in other X-linked diseases, males are affected because they only have one copy of the X chromosome. In Lesch-Nyhan syndrome, the defective gene is that for hypoxanthine-guanine phosphoribosyltransferase (HPRT), a participant in the purine metabolism. Female carriers have a second X chromosome, which contains a "normal" copy of HPRT, preventing the disease from developing, though they may have increased risk of hyperuricemia.
Formation of DNA (during cell division) requires nucleosides, molecules that are the building blocks for DNA. The purines (adenine and guanine) and pyrimidines (thymidine and cytosine) are bound to deoxyribose and phosphate and incorporated as necessary. Normally, the nucleosides are synthetized de novo from amino acids and other precursors. A small part, however, is generated from the degraded DNA of broken-down cells. This is termed the "salvage pathway".
HPRT is the "salvage enzyme" for the purines: it channels adenosine (in its hypoxanthine form) and guanine back into DNA synthesis. Failure of this enzyme has two results:
- Cell breakdown products cannot be reused, and are therefore degraded. This gives rise to increased uric acid, a purine breakdown product.
- The de novo pathway is stimulated due to an excess of PRPP (5-phospho-D-ribosyl-1-pyrophosphate or simply phosphoribosyl-pyrophosphate).
It is not known whether the neurological abnormalities in LNS are due to uric acid neurotoxicity or to a relative shortage in "new" purines during essential steps. Polymorphisms for enzymes in the de novo pathway may contribute to the disease, but this would not be the case if uric acid neurotoxicity were the main cause of the symptoms.
Various mutations of HPRT are known. Mutations that only mildly decrease the enzyme's function do not normally cause LNS, but do increase susceptibility to gout and nephrolithiasis.
Oxidative stress[]
Significantly, uric acid is a powerful reducing substance. As such, it is the single most important extracellular antioxidant, accounting for roughly half of the antioxidant ability of plasma. It may even partially substitute for ascorbic acid in human evolution.
However, like most strong reducing agents, uric acid can also act as a prooxidant, particularly at higher concentrations like those in LNS. Further, oxidation of purines by Xanthine oxidase to uric acid produces reactive oxygen species. Thus, free radicals, oxidative stress, and reactive oxygen species may play some role in the etiology of Lesch-Nyhan's syndrome [1]. Such processes also likely figure in other hyperuricemia syndromes such as that in dalmatian dogs, which responds to "Orgotein", a veterinary formulation of bovine liver superoxide dismutase. There is additional experimental support for oxidative stress in LNS. E.g., here,here and here.
Likewise, strong reducing substances such as uric acid form Charge transfer complexs with neuromelanin in pigmented midbrain structures such as the substantia nigra and the locus ceruleus. The role of this in LNS is unknown, but it might be related to (e.g.) the extrapyramidal symptoms.
Treatment[]
Treatment for LNS is symptomatic. Gout can be treated with allopurinol to control excessive amounts of uric acid. Kidney stones may be treated with lithotripsy, a technique for breaking up kidney stones using shock waves or laser beams. There is no standard treatment for the neurological symptoms of LNS. Some may be relieved with the drugs carbidopa/levodopa, diazepam, phenobarbital, or haloperidol.
It is essential that the overproduction of uric acid be controlled in order to reduce the risk of nephropathy, nephrolithiasis, and gouty arthritis. The drug allopurinol is utilized to stop the conversion of oxypurines into uric acid, and prevent the development of subsequent arthritic tophi (produced after having chronic gout), renal stones]] (also known as kidney stones), and nephropathy, the resulting kidney disease. Allopurinol is taken orally, at a typical dose of 3-20 mg/kg per day. The dose is then adjusted to bring the uric acid level down into the normal range (<3 mg/dL). Most affected individuals can be treated with allopurinol all through life.
No medication is effective in controlling the extrapyramidal motor features of the disease. Spasticity however can be reduced by the administration of baclofen or benzodiazepines.
No method of treatment for the neurobehavioral aspects of the disease has been effective. Even children treated from birth with allopurinol develop behavioral and neurologic problems, despite never having had high serum concentrations of uric acid. Self-injurious and other behaviors are best managed by a combination of medical, physical, and behavioral interventions. The self-mutilation is often reduced by using restraints. Sixty percent of individuals have their teeth extracted in order to avoid self-injury, which families have found to be an effective management technique. Because stress increases self-injury, behavioral management through aversive techniques (which would normally reduce self-injury) actually increases self-injury in individuals with LNS. Nearly all affected individuals need restraints to prevent self-injury, and are restrained more than 75% of the time. This is often at their own request, and occasionally involves restraints that would appear to be ineffective, as they do not physically prevent biting. Families report that affected individuals are more at ease when restrained.
Prognosis[]
The prognosis for individuals with LNS is poor. Death is usually due to renal failure in the first or second decade of life.
History[]
Michael Lesch was a medical student at Johns Hopkins Hospital, where pediatrician Bill Nyhan was a faculty member, when the two identified LNS and its associated hyperuricemia in two affected brothers, ages 4 and 8.[1] Lesch and Nyhan published their findings in 1964.[2] It took only three years until the metabolic cause was identified by Jay Seegmiller and his colleagues at NIH.[3]
Notes[]
- ↑ Nyhan WL. The recognition of Lesch-Nyhan syndrome as an inborn error of purine metabolism. J Inher Metab Dis 1997;20:171-8. PMID 9211189.
- ↑ Lesch M, Nyhan WL. A familial disorder of uric acid metabolism and central nervous system function. Am J Med 1964;36:561-70. PMID 14142409.
- ↑ Seegmiller JE, Rosenbloom FM, Kelley WN. Enzyme defect associated with a sex-linked human neurological disorder and excessive purine synthesis. Science 1967;155:1682-4. PMID 6020292.
References[]
- OMIM 308000 and OMIM 300322
- Cleveland clinic
- Joseph F. Smith medical library
- Overview of condition at NLM Genetics Home Reference
External links[]
- NIH/NINDS Lesch-Nyhan Information Page
- CLIMB (Children Living with Inherited Metabolic Diseases)
- Lesch-Nyhan Syndrome Registry
- The Arc (a national organization on intellectual disability)
- National Organization for Rare Disorders (NORD)
- National Institute of Child Health and Human Development (NICHD)
- Philip Baker's Homepage
- "Free Radicals and Human Disease, a Review".
amino-acids Phenylketonuria - Alkaptonuria - Ochronosis - Tyrosinemia - Maple syrup urine disease - Propionic acidemia - Methylmalonic acidemia - Isovaleric acidemia - Primary carnitine deficiency - Cystinuria - Cystinosis - Hartnup disease - Homocystinuria - Citrullinemia - Hyperammonemia - Glutaric acidemia type 1
carbohydrates Lactose intolerance - Glycogen storage disease (type I, type II, type III, type IV, type V), Fructose intolerance, Galactosemia
Lipid storage disorders Gangliosidosis - GM2 gangliosidoses (Sandhoff disease, Tay-Sachs disease) - GM1 gangliosidoses - Mucolipidosis type IV - Gaucher's disease - Niemann-Pick disease - Farber disease - Fabry's disease - Metachromatic leukodystrophy - Krabbe disease - Neuronal ceroid lipofuscinosis - Batten disease - Cerebrotendineous xanthomatosis - Wolman disease - Cholesteryl ester storage disease
List of fatty acid metabolism disorders - Hyperlipidemia - Hypercholesterolemia - Familial hypercholesterolemia - Xanthoma - Combined hyperlipidemia - Lecithin cholesterol acyltransferase deficiency - Tangier disease - Abetalipoproteinemia
mineral metabolism Disorders of calcium metabolism - Hypophosphatemia - Hypophosphatasia - Wilson's disease - Menkes disease - Hypermagnesemia - Hypomagnesemia - Hypercalcaemia - Hypocalcaemia
fluid, electrolyte and acid-base balance Electrolyte disturbance - Hypernatremia - Hyponatremia - Respiratory acidosis - Metabolic acidosis - Lactic acidosis - Hypervolemia - Hypokalemia - Hyperkalemia - Mixed disorder of acid-base balance - Hyperchloremia - Hypochloremia - Dehydration
porphyrin and bilirubin Acatalasia - Gilbert's syndrome - Crigler-Najjar syndrome - Dubin-Johnson syndrome - Rotor syndrome - Porphyria (Acute intermittent porphyria, Gunther's disease, Porphyria cutanea tarda, Erythropoietic protoporphyria, Hepatoerythropoietic porphyria, Hereditary coproporphyria, Variegate porphyria)
glycosaminoglycan Mucopolysaccharidosis - Hurler syndrome - Hunter syndrome - Sanfilippo syndrome - Morquio syndrome
glycoprotein I-cell disease - Pseudo-Hurler polydystrophy - Aspartylglucosaminuria - Fucosidosis - Alpha-mannosidosis - Sialidosis
other Alpha 1-antitrypsin deficiency - Cystic fibrosis - Familial Mediterranean fever - Lesch-Nyhan syndrome
| This page uses Creative Commons Licensed content from Wikipedia (view authors). |